2013: Journal of Physical Chemistry A
https://doi.org/10.1021/jp312492v
Jason K. Cooper, Christian D. Grant*, and Jin Z. Zhang*
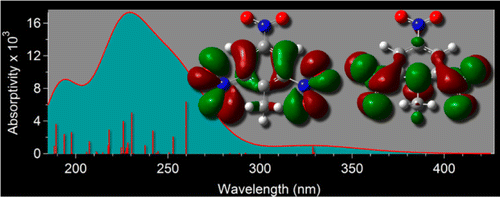
Time dependent density function theory (TD-DFT) has been utilized to calculate the excitation energies and oscillator strengths of six common explosives: RDX (1,3,5-trinitroperhydro-1,3,5-triazine), β-HMX (octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine), TATP (triacetone triperoxide), HMTD (hexamethylene triperoxide diamine), TNT (2,4,6-trinitrotoluene), and PETN (pentaerythritol tetranitrate). The results were compared to experimental UV–vis absorption spectra collected in acetonitrile. Four computational methods were tested including: B3LYP, CAM-B3LYP, ωB97XD, and PBE0. PBE0 outperforms the other methods tested. Basis set effects on the electronic energies and oscillator strengths were evaluated with 6-31G(d), 6-31+G(d), 6-31+G(d,p), and 6-311+G(d,p). The minimal basis set required was 6-31+G(d); however, additional calculations were performed with 6-311+G(d,p). For each molecule studied, the natural transition orbitals (NTOs) were reported for the most prominent singlet excitations. The TD-DFT results have been combined with the IPv calculated by CBS-QB3 to construct energy level diagrams for the six compounds. The results suggest optimization approaches for fluorescence based detection methods for these explosives by guiding materials selections for optimal band alignment between fluorescent probe and explosive analyte. Also, the role of the TNT Meisenheimer complex formation and the resulting electronic structure thereof on of the quenching mechanism of II–VI semiconductors is discussed.
