2014: Chemistry of Materials
https://doi.org/10.1021/cm5025074
Jason K. Cooper, Sheraz Gul, Francesca M. Toma, Le Chen, Per-Anders Glans, Jinghua Guo, Joel W. Ager, Junko Yano, Ian D. Sharp*
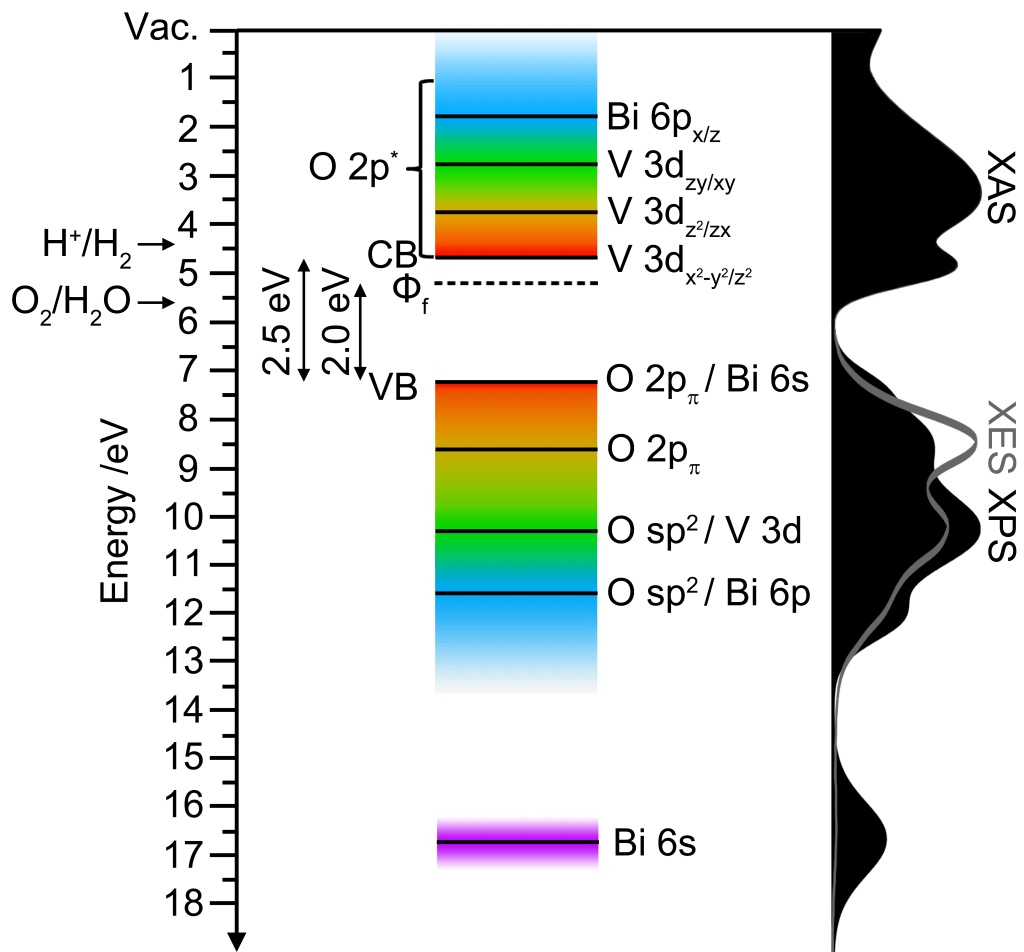
A comprehensive approach to understanding the electronic structure of monoclinic scheelite bismuth vanadate (ms-BiVO4), including both valence band (VB) and conduction band (CB) orbital character, is presented. Density functional theory (DFT) calculations are directly compared to experimental data obtained via X-ray absorption spectroscopy (XAS), X-ray emission spectroscopy, resonant inelastic X-ray spectroscopy (RIXS), and X-ray photoelectron spectroscopy to provide a complete portrait of the total and partial density of states (DOS) near the bandgap. DFT calculations are presented to confirm the VB maximum and CB minimum are comprised primarily of O 2p and V 3d orbitals, respectively. Predicted triplet d-manifold splitting of V 3d CB states, arising from lone pair-induced lattice distortions, is quantified by V L- and O K-edge XAS. Furthermore, the partial contributions to the total DOS within both the CB and VB, determined by RIXS, are found to be in excellent agreement with DFT calculations. Energy levels are placed relative to the vacuum level by photoemission spectroscopy, which provides a measure of the work function and electron affinity of the investigated BiVO4 thin film. The implications of the fundamental electronic structure of ms-BiVO4 on its photocatalytic behavior, as well as considerations for improvements by substitutional incorporation of additional elements, are discussed.
